Züchtungsinformatik: Neue Wege zur Beschreibung von Tier- & Pflanzen-Stammbäumen
Universität Hohenheim arbeitet an Verfahren, wie sich Entwicklungen im Erbgut von Tieren und Pflanzen besser nachvollziehen lassen.

Die Züchtungsinformatik beschreitet neue Wege zur Beschreibung von Tier- & Pflanzen-Stammbäumen
ClipDealer
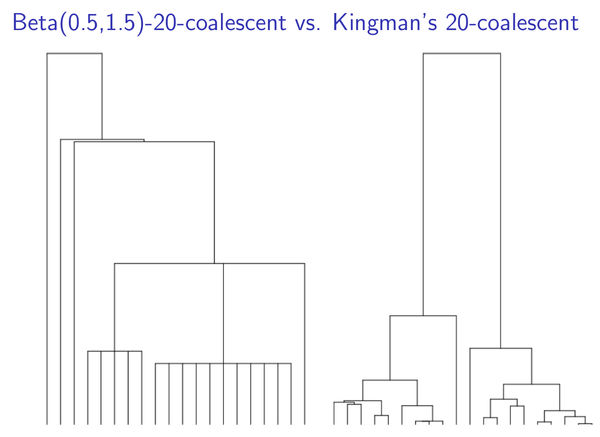
Multiple-Merger-Coalescent-Modell und Kingman-n-Coalescent-Modell im Vergleich.
Fabian Freund

Menschliche Stammbäume lassen sich mit Hilfe von Dokumenten mitunter mehrere Jahrhunderte zurückverfolgen. Bei vielen Pflanzen und Tieren sind mathematische Verfahren notwendig, die sorgfältig ausgewählt und aufwendig berechnet werden müssen. Dr. Fabian Freund von der Universität Hohenheim in Stuttgart arbeitet daran, diese Modelle zu verbessern, die Auswahl zu vereinfachen. Die Deutsche Forschungsgemeinschaft fördert das Projekt mit 258.700 Euro. Damit zählt es zu den Schwergewichten der Forschung an der Universität Hohenheim.
Die Nase vom Papa, die Augen von der Mama: Babys schreibt man gerne bestimmte vererbte Merkmale zu. Wer die Entstehung solcher Eigenschaften im Tier- und Pflanzenreich nachvollziehen will, muss meist auf mathematische Modelle zurückgreifen.
Diese Modelle beschreiben, wie der Stammbaum ausgesehen haben könnte und an welcher Stelle es bei ihm zu einer genetischen Veränderung kam. So lässt sich nachvollziehen, wie sich eine Art entwickelt hat – und das über Jahrtausende hinweg.
Wahl des richtigen Modells ist entscheidend
Dabei gilt jedoch: Stammbaum ist nicht gleich Stammbaum. Aktuell geht die Wissenschaft davon aus, dass es zwei typische Muster gibt, wie sich Stammbäume verzweigen:
-
Die Äste des Stammbaums verzweigen sich relativ oft, aber immer nur in genau zwei neue Zweige. Auf diese Weise entstehen viele Verästelungen, an deren Ende viele kleine Familien stehen. Mathematisch ist dies das sogenannte „Kingman-n-Coalescent-Modell“.
Die Äste des Stammbaums verzweigen sich seltener, dafür können sich die Äste in eine Vielzahl von Zweigen auffächern. Auf diese Weise entstehen weniger, dafür aber größere Familien mit gemeinsamem Erbgut. Die mathematische Bezeichnung dafür lautet „Multiple-Merger-Coalescent-Modell“.
Das erste Modell ist das Standard-Modell. Das Zweite gilt speziell für Populationen mit vielen Nachkommen pro Individuum oder Paar, von denen allerdings nur wenige überleben. Letzteres ist zum Beispiel für Populationen typisch, deren Umwelt sich stark verändert und deshalb einen starken Evolutionsdruck auf die Population ausübt.
Wenn Forscher einen Stammbaum durch Computer-Simulation erstellen wollen, müssen sie erst herausfinden, welcher der beiden mathematischen Ansätze der zutreffende ist.
Neues Verfahren soll Entscheidung vereinfachen
Die Antwort auf diese Frage ist allerdings zeit- und arbeitsaufwändig. Beantworten lässt sie sich bislang nur, indem man sehr viele Stammbäume aus beiden Modellen ausprobiert und die Ergebnisse mit den realen Daten vergleicht.
Dr. Fabian Freund vom Fachgebiet Nutzpflanzenbiodiversität und Züchtungsinformatik an der Universität Hohenheim arbeitet deshalb daran, den Aufwand zu verringern. Gleichzeitig soll seine Forschung noch die Treffsicherheit der Entscheidung erhöhen.
Um das zu erreichen arbeitet der Mathematiker mit einer verfeinerten Datenauswahl: „Unsere Computersimulationen haben uns gezeigt, dass wir schon dadurch eine Entscheidungshilfe bekommen, indem wir statt ganzer Stammbaum-Äste nur die Erbgut-Sequenz der kleinsten beobachtbaren Familien ablesen – quasi die Blätter am Ende des Astes.“
Multiple Merger Coalescent-Modelle auf dem Prüfstand
Dabei könnte es sogar sein, dass die Arbeit von Dr. Freund die Modellauswahl noch viel radikaler vereinfacht – indem sie das eine Modell als irrelevant entlarvt. „Es gibt den Verdacht, dass Stammbäume nach dem Multiple Merger Coalescent-Modell nur in der Theorie existieren und praktisch gar nicht – oder nur für sehr wenige Spezies – vorkommen.“
Gemeinsam mit anderen Züchtungsforschern der Universität Hohenheim hat Dr. Freund deshalb bereits damit begonnen, die Arten genauer zu untersuchen, die bislang als Vertreter von Stammbäumen nach dem Multiple Merger coalescent-Modell galten. „Erste Testrechnungen laufen für Fadenwürmer-Arten und Tuberkulose-Bakterien sowie in Zusammenarbeit mit dem Leiter des Fachgebiets Prof. Dr. Karl Schmid für einen Maispilz.“ Untersuchungen für weitere Pflanzen-, Pilz- oder Virenarten sollen folgen.
Sollte sich herausstellen, dass sich ihr Stammbaum ebenso gut durch das Kingman-n-Coalescent-Modell darstellen ließe, könnte die Auswahl zwischen verschiedenen Modellen künftig komplett entfallen.

Weitere News aus dem Ressort Wissenschaft


























































